



Retinoblastoma – Definition and Overview
Retinoblastoma is a tumor in the eye that mainly affects children below 5 years of age i.e. in early childhood. It may also develop in fetuses in the womb, as well as in newborns, babies and toddlers.
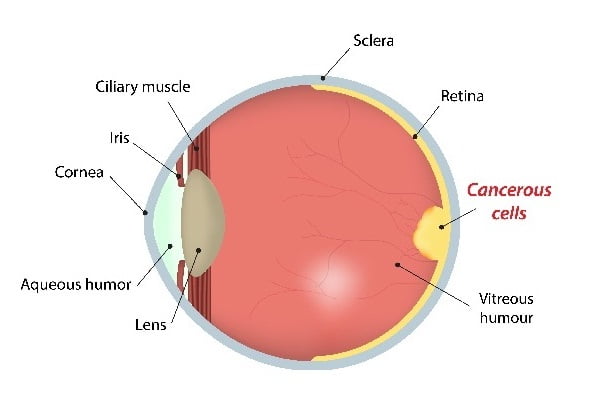
Retinoblastoma is rare malignant eye tumor. It starts in the retina (a sensitive layer of nerve cells lining at the back of the eye that enables visibility) of the eye. The nerve cells begin to grow in an uncontrolled manner making the tumor malignant.
ADVERTISEMENT
ADVERTISEMENT
Cancer Research UK defined retinoblastoma as ‘retino’ means from the retina, ‘blast’ means cells in early development phase and ‘oma’ means a group of cells, or a tumor.
Retinoblastoma is a “not so commonly” occurring form of eye cancer and may occur either in one eye (unilateral) or both the eyes (bilateral). Unilateral cases are about 60% whereas 40% of the cases are of bilateral type.
Retinoblastoma occurs approximately 1 in 15,000 to 1 in 18,000 live births. It may be inherited or not. If it is left untreated, severe complications may occur. The cancer may spread outside of the eye (into the eye socket), in the lymph nodes, bones, into the brain (rare) and eventually to other parts of the body resulting in death. Therefore, retinoblastoma is not just sight-threatening but life-threatening as well.
Approximately 9 out of 10 children with retinoblastoma can be cured. Retinoblastoma can affect everyone irrespective of gender, race, ethnicity etc. It even occurs equally in the right or left eye.
See also:
Eye Cancer (Ocular Cancer): Causes, Symptoms, Types, Stages, Risks, Treatment
Tumor Behind Eye: What Does It Mean? Is It Cancer?
A brief history of retinoblastoma
In 1597, retinoblastoma was described as a disorder by Pawius. Then, in 1809, Wardrop described the tumor as fungus hematodes, with enucleation as its primary treatment. During 1890s, Flexner and Wintersteiner stated that it was a neuroepithelioma due to the presence of rosettes. Later, it was found that the tumor originated from the retinoblasts and the American Ophthalmological Society officially accepted the term retinoblastoma in 1926.
The survival rate of patients with retinoblastoma increased from 5% in 1896 to 81% in 1967.
Two types of retinoblastoma
- Heritable retinoblastoma: Retinoblastoma is linked with familial mutations, which means mutations may occur in the reproductive cells (sperm and eggs) in the retinoblastoma-RB1 gene. It is also known as congenital retinoblastoma.
Newborns with a mutation in the RB1 gene generally develop retinoblastoma in both eyes (called bilateral retinoblastoma). Several tumors develop within the eye (called multifocal retinoblastoma) of these children. Of all the cases of retinoblastoma, about 30% are heritable retinoblastoma. - Non-heritable retinoblastoma: It is a non-familial disease which initiates from somatic (from non-reproducing cells) mutations. It is supposed to be sporadic in nature. This means it can occur without any family history. The mutations in the RB1 gene shall not pass to future generation. Approximately, 80% of the cases are non heritable retinoblastoma. The exact reason for why non-heritable retinoblastoma occurs is still unknown.
The over-grown cells form tumors on the retina or may float through the vitreous humor to other parts of the eye resulting in the formation of more tumors. The pressure inside the eye increases as tumors block the channels that let the fluid circulate inside the eye. This sometimes leads to glaucoma i.e. pain and loss of vision in the affected eye.
Retinoblastoma in children
Chromosomes have genetic codes which control the growth and development of the cells. 80% of retinoblastoma cases occur without any former indication and disorder, and the rest of about 20% have a family history with retinoblastoma. Retinoblastoma is very rarely caused by external factors such as smoking, drinking, etc.
There is a genetic link to retinoblastoma. Kids who carry the genetic mutation usually get more than one tumor and are likely to develop the disease in both eyes. The disease is known as a childhood disease.
What age does retinoblastoma occur?
Retinoblastoma occurs mostly in children under the age of 4 years, with about 80% of cases occurring under age 3 years and almost none above age 6 years. Most of the children (about 99%) with retinoblastoma are cured of the disease.
Possible risk factors for retinoblastoma
Some known risk factors for retinoblastoma are:
Age
Retinoblastoma is generally diagnosed in children below 5 to 6 years of age. Most of the times, congenital or hereditary retinoblastoma is detected in the first year of a child’s life and the non-inherited retinoblastoma tend to be diagnosed in children below 2years of age.
Genetics and heredity
Mainly, the genetic mutations or changes in the human body can lead to retinoblastoma. It is caused by mutations in RB1 gene in retinoblasts. The RB1 gene has 2 copies, one in each parent and changes occur in both the copies of RB1 gene for the retinoblastoma tumor to develop. Genetic mutations are responsible for about 40% of retinoblastoma cases, almost all of them in children.
About 1 in 4 retinoblastoma cases is inherited from the parent. In the rest, gene mutation is not inherited, but occurs during an early development of a child in the womb. Inherited mutation in the RB1 gene usually causes bilateral retinoblastoma. Only about 15% to 20% of the children suffering with retinoblastoma have a family history of the disease.
The chances of developing other types of cancer also increase in children who have had bilateral retinoblastoma or the hereditary form of unilateral retinoblastoma. A few of such cancer types that can occur in children are:
- bone cancer
- soft tissue sarcoma
- melanoma skin cancer
- brain cancer
- lung cancer
Amplification of the MYCN gene: Though rarely, but another gene known as MYCN gene amplifies at a significant rate, forming large tumors in one eye leading to retinoblastoma. It is more common in children younger than 6 months in age. It cannot be passed on to children and does not increase the risk of developing other cancers.
MYCN gene functions in making a protein which plays a significant role in the formation of tissues and organs during embryonic development.
Although the risk of retinoblastoma is raised by genetic mutation, but it doesn’t mean that genetics is the only cause of retinoblastoma.
What causes retinoblastoma?
Mostly, RB1 gene suffers mutation and causes retinoblastoma in children. Family history may also be responsible for retinoblastoma in some cases if the cancer occurs at a younger age without any mutation. This type of cancer is called hereditary retinoblastoma.
What is RB1 gene?
The function of RB1 gene is to provide instructions for the production of a protein called pRB. pRB is the retinoblastoma gene which acts as a tumor suppressor located on chromosome 13q14. It regulates cell growth by inhibiting the cell cycle. This maintains the rate of cell division and prevents the cell from dividing too quickly in an uncontrolled way.
Under specific conditions, pRB stops other proteins from triggering DNA replication (to make a copy of the DNA). This in turn controls cell division and prevents the growth of tumor. It is essential that this process occurs before a cell can divide.
How does RB1 gene work for retinoblastoma?
RB1 gene produces a protein (pRb) which hinders the excessive and fast growth of cells. An individual cell has 2 RB1 genes and even if any one of them works, retinoblastoma does not occur. If both the RB1 genes are mutated and missing, a cell grows unchecked which leads to further gene changes, which in turn may cause cells to become cancerous.
A single copy of the mutated RB1 gene in a single cell is sufficient to cause hereditary or bilateral retinoblastoma. This is an autosomal dominant pattern. The faulty gene is either inherited from a parent, or originates through a change in the gene (mutation) at an early stage of the child’s development in the womb.
In non-hereditary (sporadic) retinoblastoma, there is no family history of the disease. The abnormality in the gene develops on its own in only one cell in one eye. Scientists do not know what exactly causes this change on its own. A child who has sporadic (non-hereditary) retinoblastoma develops only one tumor in one eye.
People with non-hereditary retinoblastoma are not at risk of passing RB1 gene mutations to their children.
How can retinoblastoma be prevented?
There’s no proven way to prevent retinoblastoma as there are no known avoidable risk factors. Smoking (especially during pregnancy) and exposure to chemicals in the workplace should be avoided. When the tumor is small, many preventive measures can be considered for child so that the tumor might not grow. If the tumor still grows, retinoblastoma can be cured at an initial stage.
Prevention for families with inherited retinoblastoma
Family history should be known before the birth of a child to provide proper cure or preventive measures if there is a risk of getting retinoblastoma. For early diagnosis of retinoblastoma, there are a few genetic tests that enable families to identify whether their children are at an increased risk of retinoblastoma or not.
Children whose parents have a history of retinoblastoma are recommended to be screened before and right after their birth for an early detection of the cancer. Early detection of this cancer improves the chance for successful treatment significantly. Genetic counseling can also be a way of creating awareness about the heritable retinoblastoma to lower the risk of gene passing to the next generation.
Early detection and screening for retinoblastoma: Is there a way to detect retinoblastoma early?
Retinoblastoma is a rare cancer which is found in children below 5 years of age. It shows no specific symptoms and no screening tests are therefore recommended for retinoblastoma in children.
Affected parents should be aware of the symptoms of retinoblastoma in their children (as it is likely to be inherited). Routine checkups of eye reduce the risk to some extent or may help detect it in early stage, which can be treated better. Children with affected RB1 gene or with a family history of retinoblastoma can suffer with retinoblastoma during their initial stages of life.
Tumors can be detected in eyes at an early stage through an eye test. A special blood (genetic) test for newborns helps in diagnosing the defect in RB1 gene during the first few weeks’ after the birth.
If tumor occurs in both eyes, it generally happens at the same time. MRI scans of the brain are recommended at regular intervals to see if retinoblastoma is developing.
Symptoms and signs of retinoblastoma
The signs and symptoms of retinoblastoma may include:
Leukocoria
A white color reflection occurs in the center circle (pupil) of the eye when exposed to light, such as while taking a flash photograph. It is called leukocoria or cat’s eye reflex or white pupillary reflex. In normal conditions, the pupil appears to be red in color in flash light due to the presence of blood vessels at the back of the eye.
Strabismus
Sometimes, eyes appear to be looking in different directions or are crossed. It is known as strabismus (“wandering eye”). This condition is called “lazy eye condition”. Strabismus is caused by a mild weakness of the muscles that control the eyes. Lazy eye can also be caused by retinoblastoma.
Glaucoma
Another symptom for retinoblastoma is an increased pressure in the eye which can often lead to blindness. This condition is known as Glaucoma.
Proptosis
The bulging of the eye out of the eye socket is called proptosis. This can also cause retinoblastoma.
Heterochromia
A condition in which two-different colored irises appear is called heterochromia. This may be a sign of retinoblastoma.
Other symptoms for retinoblastoma are:
- Swelling in the eye or an enlarged eyeball
- Squint eye
- Redness in the white part of the eye (sometimes inflammation also occurs)
- Loss of or blurred vision
Retinoblastoma diagnosis
Diagnosis of retinoblastoma usually begins with common eye tests when unusual signs and symptoms are visible and a loss in the vision of the eye is reported.
Following tests are done for diagnosing retinoblastoma:
Physical examination and medical history for retinoblastoma
Generally, diagnosis of retinoblastoma is done when signs and symptoms start appearing. The symptoms are observed by an eye specialist carefully. He or she then proceeds with (physical) examination of the eye. Ophthalmoscope, slit lamp and optomap are devices that are used during physical examination of eyes.
Another type of eye exam is known as EUA (examination under anesthesia). During ‘Examination under Anesthesia’, the size, location and shape of the tumor inside the eye is photographed. These pictures map out the exact problem in the eye.
Hereditary and family history is an important factor in retinoblastoma. Family history also helps to see through if other relatives of the family have also suffered with changes in RB1 gene which can lead to retinoblastoma.
Imaging tests for retinoblastoma
Tests used for imaging mechanize x-rays, sound waves, magnetic fields, or radioactive substances to generate pictures of the inside of the body. Imaging tests are done for confirming a doubtful retinoblastoma. Imaging tests are conducted:
- To detect any tumor in the eye which can be a reason for retinoblastoma
- To detect the size of a tumor and its metastasis
- To look for an apt treatment to cure retinoblastoma
Ultrasound for retinoblastoma
Sound waves are used to create images of the tissues inside the body. This is a painless test. The tumor produces different echoes of the sound waves. Sound waves bounce back as echoes and forms images on the screen after being converted into signals. An ultrasound probe is used to detect the tumor during ultrasound. Ultrasound helps in detection of large tumors and is not harmful for a child as there are no harmful radiations involved.
Ultrasound is one of the most common imaging tests for the diagnosis of retinoblastoma. This test can be very useful when tumors in the eye are so large that they prevent the doctors from seeing inside the whole eye. The probe is gently placed over the closed eyelids or sometimes directly on the surface of the eye. Anesthetic eye drops are used to create numbness in the eye before an ultrasound is done.
Magnetic resonance imaging (MRI) scan for retinoblastoma
Strong magnetic forces and radiofrequency waves help in detection of the size and number of tumors in the eye. Magnetic resonance imaging (MRI) uses a 3D imaging system which provides a cross-section of the inside of the eye (the blood vessels, tissues, the optic nerve, etc).
If the tumor accumulates in both the eyes of a child (bilateral retinoblastoma), then after several years, there is a great possibility that the tumor may hit the pineal gland in the brain (trilateral retinoblastoma) which can be detected by an MRI scan of the brain.
Gadonium is a contrast material which is injected into a vein before the MRI scan to provide detailed pictures to the affected area.
CT scan for retinoblastoma
CT scan mechanizes X-rays to produce detailed images of the tumor. A CT scan can increase the risk of cancer in children because the mechanism of CT scan depends on the use of harmful radiations. The diagnosis of retinoblastoma is clearer with CT scan than that with MRI.
A CT scan shows deposits of calcium in the tumor much better than an MRI. This helps in clearer diagnosis of retinoblastoma. Sometimes, serious reactions like trouble breathing, flushing (a warmth feeling on the face), low blood pressure or allergy might occur in children after CT scan due to the dye which is given as an intravenous injection. The contrast dye helps to significantly outline structures in the body.
Bone scan and biopsy plus bone marrow aspiration
Radio-pharmaceuticals (bone-seeking radioactive materials), with the help of a computer, are used to take pictures of areas of bones and skull where the cancer has metastasized.
Sometimes, two tests, biopsy and bone marrow aspiration, are done together to check if the cancer has or not metastasized to the bone marrow (the soft inner part of certain bones). These tests are usually not needed unless the retinoblastoma has grown outside the eye. A bone marrow biopsy is usually done just after the aspiration.
Other tests for retinoblastoma
RB1 gene test
RB1 stands for retinoblastoma 1. RB1 test is done to check for mutations in the genes during retinoblastoma. A blood or tissue sample is collected and tested in labs to study the mutations, if, occurring in the RB1 gene.
Lumbar puncture (spinal tap) for retinoblastoma
Retinoblastomas can sometimes grow along the optic nerve and extend to the brain surface. The cancerous cells can often mobilize in the cerebrospinal fluid (surrounding the brain and the spinal cord) for extension.
A small, hollow needle inserted between the bones of the spine is used to withdraw a small amount of fluid which is tested in a lab to detect cancerous cells.
Stages of retinoblastoma
The stage of a disease classifies the extent of spread of the disease. Different stages can provide information about change in size or type of the tumor. Most importantly, stage distinction helps in choosing the right method of treatment.
Retinoblastomas are divided into 2 main groups:
- Intraocular retinoblastoma: The tumor is still supposed to be inside the eye in case of intraocular retinoblastoma. Either in 1 or both eyes, intraocular retinoblastoma does not extend to the surrounding tissues or to other parts of the body. More than 9 out of 10 (about 90%) of the children affected with intraocular retinoblastoma can be cured.
- Extraocular retinoblastoma: The cancer which extends beyond the eyes is extraocular retinoblastoma. Extraocular retinoblastoma can be further classified into orbital retinoblastoma (which extends only to the eye socket) and metastatic retinoblastoma, which spreads its roots to distant parts of the body, such as the brain or bone marrow.
The staging of retinoblastoma through some detailed systems
New researches and clinical trials have shown a positive way for the treatment of different cancer types. Defining a stage is very important for treating retinoblastoma.
International classification system for intraocular retinoblastoma
The International Classification for Intraocular Retinoblastoma is a brand new staging system, which uses “groups” to provide an estimate of the eye condition. It provides all the details of the disease from recent decades. Intraocular retinoblastoma is divided into 5 groups, labeled A to E, based on the possibility of retaining vision using the latest treatments.
Group A retinoblastoma
In group A retinoblastoma, tumors are present only in the retina and are of small sizes (3 millimeters [mm] across or less). These tumors do not appear near the important structures of the eye such as the optic disc (where the optic nerve enters the retina and leaves the back of the eye) or the foveola.
Group B retinoblastoma
Group B has all the other tumors (large or small) that are still in any part of the retina. These tumors are close to the optic disc or foveola).
Group C retinoblastoma
In group C, “seeding” occurs in some very small areas. Seeding refers to the spreading of some well defined tumors. If it is under the retina, it is called as “subretial seeding” and if the tumors spread into the jelly like material in the eye, it is termed as “vitreous seeding”.
Group D retinoblastoma
In group D, the cancer spreads at the back of the eye. The retina might detach from the back of the eye. This might lead to massive vitreous/sub-retinal seeding.
Group E retinoblastoma
The vision is lost with cancer due to the following reasons:
- The tumor has touched the lens or it has grown in the front of the eye
- The tumor can cause increased (high) pressure in the eye, known as glaucoma
- There is blood appearing in the front of the eye (hyphema) or the tumor results in bleeding in the eye (vitreous hemorrhage)
- The eye looks infected but actually it is not
Reese-Ellsworth staging systems
Developed in the 1960s, Reese-Ellesworth staging system was effective in evaluating that the child suffering with retinoblastoma will gain or lose the vision during and/or after the treatment. This system is not used commonly but sometimes it is needed to define the retinoblastoma that has not spread beyond the eye.
In early days, it was more common to treat retinoblastoma with radiation therapy (EBRT) instead of surgery to remove the eye (enucleation).
The increasing group number, from 1 to 5, lowers the chance of controlling retinoblastoma or preserving the vision of the eye:
Group 1 (very favorable for saving the vision)
1A: A single tumor which is present at or behind the equator is smaller than 4 disc diameters (DD),
1B: All multiple tumors are seen at or behind the equator. They are smaller than 4 DD
Group 2 (favorable for saving the eye or preserving)
2A: Only a single tumor which is present at or behind the equator is 4 to 10 DD in size
2B: Multiple tumors are present at or behind the equator. At least one of them is of 4 to 10 DD.
Group 3 (a doubtful condition for saving the eye)
3A: tumor of any size occurs in front of the equator
3B: a single tumor is present behind the equator and is larger than 10 DD
Group 4 (unfavorable for saving the eye)
4A: multiple tumors occur in which some are larger than 10 DD in size
4B: a tumor can enlarge and extend towards the front of the eye to the front edge of the retina known as the ora serrata.
Group 5 (very unfavorable for saving the eye)
5A: tumors cover the maximum parts of the retina
5B: during vitreous seeding, the tumor expands into the vitreous gel (jelly-like material) present the eye.
TNM staging system
Some other systems for staging retinoblastoma include both intraocular retinoblastomas and extraocular retinoblastomas. American Joint Commission on Cancer (AJCC) staging system is one such staging system. AJCC is referred as the most common and standard staging system used for all types of cancers.
The TNM staging system is mainly used to stage advanced cancers with many solid tumors that form lumps. The extent of retinoblastoma that has spread beyond the eye is also defined through TNM staging system, in detail.
It takes into account 3 key pieces of information:
T: The size of the main tumor is calculated. The growth rate is also observed within and outside of the eye
N: The cancer may or may not reach the lymph nodes.
M: The cancer may or may not have metastasized to distant parts of the body, such as the bone marrow, brain, skull, or long bones
The TNM system for staging retinoblastoma is divided into 4 stages. Generally, as the stage progresses, the risk of severity of cancer also increases.
Stage 1
The tumors can be found only in the eyeball.
Stage 2
The tumor expands to nerve or the tissues which surround the eye.
Stage 3
Retinoblastoma expands outside the eye, to the nerves and lymph nodes.
Stage 4
Cancer spreads to other parts of the body (brain, bones or lungs) at this stage. This expansion is known as distant metastasis. This type of retinoblastoma is known as metastatic retinoblastoma.
An additional staging system for the spread of cancer beyond the eye is Extraocular staging system:
Stage 0 (zero) Retinoblastoma
Stage 0 signifies deals with intraocular cancer i.e. retinoblastoma has not spread outside the eye (patients with stage 0 disease are classified based on the International Classification System, mentioned above). No additional (adjuvant) treatment is required after surgery at this stage.
Stage 1 Retinoblastoma
At this stage, the eye has already been removed and there is minor growth of cancer in the optic nerve. Patients of this stage require adjuvant chemotherapy after the surgery.
Stage 2 Retinoblastoma
Tumor spreads to the optic nerve or the sclera. Adjuvant chemotherapy and possibly radiation therapy is required at this stage.
Stage 3 Retinoblastoma
Retinoblastoma extends to the lymph nodes or the bone cavity surrounding the eyeball.
This stage is divided into 2 stages (a and b) depending on the area where the tumor extends. Both chemotherapy and radiation therapy are used as treatment.
Stage 4 Retinoblastoma
In the final stage, the tumor expands to distant areas of the body outside the eye, via the lymphatic system and blood vessels. High dose of chemotherapy with stem cell therapy or bone marrow transplantation is the treatment methodology for this stage.
The old worn out bone marrow cells are replaced by new healthy cells. Radiation therapy is sometimes combined with high-dose chemotherapy during the treatment.
Stage 4 Retinoblastoma is subdivided into group A and B depending upon the location where the cancer has spread.
Treatment of Retinoblastoma
As a rare disease, Retinoblastoma requires special treatments than any other eye disorder (especially in children). About 90% of the cases of retinoblastoma can be cured and there is a possibility of long term survival.
Principle factors considered during the treatment of retinoblastoma
Position of the tumor (in one or both eyes), vision quality, growth and expansion of the tumor outside the eye are some key points which set a base for the treatment of retinoblastoma.
The aims ensured during the treatment of retinoblastoma are:
- Preservation of the vision
- Save the eye as far as possible
- Removal of eye and saving the life of the child
- The risk of metastatic cancer is reduced
- Reduce the chances for second cancer in children (in case of hereditary retinoblastoma)
Treatment methodologies for Retinoblastoma
Surgery or Enucleation for Retinoblastoma
Surgery refers to the complete removal of a tumor (affected part) with some healthy tissues surrounding it. Surgery is only preferred in case of extended (large) tumors in the eye. There are minor chances of saving the vision if the tumor spreads in the surrounding tissues. Enucleation is an operation where the whole eye along with the attached optic nerve is removed.
An implant made up of silicone or hydroxyapatite (a material similar to bone) is used after the surgery. The implant functions same as the eye would have, naturally and is attached to the muscles in the eye socket. A few weeks after the surgery, an artificial eye (customized) can be made and clipped onto the eye implant right behind the eyelids (just like the natural eye). Again, this eye results in no visibility but it appears like a healthy eye.
Side effects of surgery for retinoblastoma
- The most common side effect of enucleation is vision loss in one eye or in both eyes (although in most cases the vision has already been lost).
- The growth of bone and surrounding tissues in the eye socket are affected.
Radiation Therapy for Retinoblastoma
Radiation therapy uses X-rays to destroy cancer cells and is an effective treatment for some children with retinoblastoma. In comparison with surgery, there are chances to save vision with radiation therapy.
To destroy the tumor cells, in some cases, a small disk of radioactive material is placed in or around the tumor and is left for a specific duration. While in other cases, a large machine can be used to emit radiations which target the tumor from outside the body.
Types of radiation therapy used in treating retinoblastoma
Internal radiation therapy (brachytherapy)
Internal radiation therapy is also known as episcleral plaque radiotherapy. The usage of plaque brachytherapy is confined for small tumors. Here, the radioactive material is placed (sewn) directly on the eye or specifically outside the part of the eyeball for several days. The radiations emitted travel a very short distance and therefore most of them are focused only on the tumor.
“Plaque” is a small radioactive material carrier, made up of gold or lead to shield nearby tissues from the radiation.
The medium or small-sized tumors have no response to other local therapies (such as cryosurgery, thermotherapy or laser surgery), therefore internal radiation therapy is preferably used.
External beam radiation therapy
External beam radiation therapy focuses radiation beams directly on to the delicate parts of the eye and these beams are strong enough to destroy the tumor during retinoblastoma.
External beam radiation therapy is not recommended for the treatment of retinoblastoma in children due to its side effects and increased risk of developing a second cancer. It may be used during advanced stages of retinoblastoma if other treatments do not respond.
Some new forms of external beam radiation therapy are:
- Intensity modulated radiation therapy (IMRT): During IMRT, the radiation beams are shaped and focused at specific angles with calculated intensity to destroy the tumor cells.
- Proton beam therapy: Protons, which are the positive parts of atoms, are used for external radiation therapy because they cause less damage to surrounding tissues as they directly hit the tumor.
Side effects of radiation therapy
- The main side effect of radiation therapy is the damage to the retina or optic nerve, which can affect the vision later.
- The bone growth in children may lower down.
- Brachytherapy does not increase the risk of developing a second cancer but IMRT does.
- Short-term side effects such as mild sunburn like skin reactions and hair loss might occur.
Laser Therapy (Photocoagulation) for Retinoblastoma
A powerful laser beam is focused through the pupil on the blood vessels that surround the tumor. The blood vessels supply oxygen and nutrients to the tumor. Heat (caused by the beams) destroys the cancer cells and the surrounding blood vessels. It can also be used for tumor that is left behind even after chemotherapy. It is also an effective method to treat retinoblastoma that comes back within the eye.
Photocoagulation is effective only for small tumors at the back of the eye.
Side effect of photocoagulation
- Laser therapy can damage the retina and results in blind spots. It can also cause the detachment of retina temporarily from the back of the eyeball.
Cryosurgery for retinoblastoma
Cryosurgery is very commonly used for the treatment of intraocular retinoblastoma (not spread beyond the eye).
A small metal probe (cooled to very low temperatures) is placed on the outer surface of the eyeball near the tumor. Cryosurgery is the process of freezing and thawing of cancerous (retinoblastoma) cells which causes them to die.
It is an effective way to treat small tumors towards the front of the eye. It is not used for children with multiple tumors.
Possible side effects of cryotherapy
- Cryotherapy leads to inflammation or swelling of the eye and eyelid for few days.
- Similar to laser therapy, cryotherapy can damage the retina, causing blind spots or temporary retinal detachment.
Chemotherapy for Retinoblastoma
Chemotherapy involves the use of anti-cancer drugs for the treatment of retinoblastoma. It helps to shrink the tumor and is done usually with another treatment (such as radiation therapy, cryotherapy, thermotherapy or laser therapy) to destroy the remaining cancerous cells.
There are different ways in which chemotherapy can be given to a child:
- Systemic chemotherapy: The most common method of performing chemotherapy is that the drugs are injected into a vein or are given by mouth. These drugs then flow through the bloodstream and spread all over the body and kill the cancer cells.
- Periocular (subtenon) chemotherapy: In periocular chemotherapy, higher doses of chemo drugs are injected into the tissues around or inside the eye. Carboplatin (Paraplatin, Paraplatin AQ) is the most common chemotherapy drug used in periocular chemotherapy.
- Intra-arterial chemotherapy: A thin catheter is inserted into a large artery on the inner thigh. This long flexible catheter moves up to the ophthalmic artery (the main artery which carries blood to the eye) and chemotherapy drugs are infused through this artery.
Small doses of chemo drugs (less than 10%) are used in intra-arterial chemotherapy. Therefore, there are fewer side effects of this chemotherapy. Chemotherapy drugs which are most commonly used for intra-arterial chemotherapy are melphalan (Alkeran), carboplatin and topotecan (Hycamtin).
- Intravitreal chemotherapy: Chemotherapy drugs are injected into the jelly-like substance in the eye called the vitreous humour. It is supposed to be a new treatment method.
- Intrathecal chemotherapy: Intrathecal chemotherapy is mainly given to the brain and spinal cord (called the central nervous system, or CNS) through a lumbar puncture into the space containing the cerebrospinal fluid (CSF). Intrathecal chemotherapy is mainly used to treat retinoblastoma that extends to the brain.
Some drugs used during chemotherapy for retinoblastoma
Some of the drugs used to treat children with retinoblastoma include:
- Carboplatin
- Cisplatin
- Vincristine(Oncovin)
- Etoposide(Vepesid, VP-16)
- Cyclophosphamide
- Topotecan
- Doxorubicin(Adriamycin)
- Teniposide (Vumon)
- Cyclophosphamide (Procytox)
- Melphalan
The most common chemotherapy drug combinations used to treat retinoblastoma are:
- Carboplatin, Vincristine and Etoposide
- Cyclophosphamide and Vincristine
- Doxorubicin and Cyclophosphamide
- Vincristine, Carboplatin, Teniposide, and Cyclophosphamide
- Cyclophosphamide, Doxorubicin and Vincristine
- Topotecan and Cytarabine (Cytosar)
- Carboplatin, Vincristine, Etoposide and Cyclosporine (Neoral)
Side effects of chemotherapy
- Chemo drugs sometimes work against the normal cells which are present in the bone marrow (where new blood cells are made), the lining of the mouth and intestines, and the hair follicles. This happens because the cells of the bone marrow have a tendency to divide quickly like the cancerous cells.
- The use of Cisplatin and carboplatin might affect the kidney.
- Vincristine can damage the nerves and can also cause tingling and numbness, particularly in the hands and feet of children.
- Some drugs, such as doxorubicin, etoposide, cyclophosphamide, etc can increase the risk of developing a cancer of white blood cells, known as acute myeloid leukemia (AML).
- Cyclophosphamide damages the bladder, which can cause blood in the urine.
Common side effects of chemotherapy drugs used for retinoblastoma are:
- Loss of hair
- Appetite loss
- Redness, swelling or inflammation
- Fatigue
- Mouth sores
- Nausea and Vomiting
- Bone marrow suppression (low blood cell counts)
- Diarrhea and Constipation
- High risk of infections
- Frequent bleeding (low platelet count)
Thermotherapy for Retinoblastoma
Thermotherapy is also called transpupillary thermal therapy or TTT where laser (or infrared light) destroys tumor cells with heat. The temperature used is not as extremely high as that used in photocoagulation therapy. Therefore, there might remain some spared blood vessels on the retina.
For large tumors, TTT is used in combination with chemotherapy (called thermo-chemotherapy) or with radiation therapy (called thermo-radiotherapy). Heat helps these other treatments work well for some cases.
Possible side effects of thermotherapy
- Thermotherapy causes the colored part of the eye, known as the iris, to shrink.
- Thermotherapy can often lead to the clouding of a part of the eye lens or might cause damage to the retina, which in turn might affect the vision.
High-Dose Chemotherapy and Stem Cell Transplant for Retinoblastoma
Higher dose of chemotherapy is used during a stem cell transplant (SCT) in this combination treatment. This is a new treatment technology and doctors are studying its use in children with retinoblastoma that has spread outside the eye and who are unlikely to benefit from other treatment options. Earlier, this treatment was known as a bone marrow transplant.
For Stage 4 extraocular-retinoblastoma patients, a stem cell transplant may be recommended.
Sometimes, healthy stem cells are collected from the bone marrow of the child (autologous stem cells) before high-dose chemotherapy and stored. After the chemotherapy session, these cells are injected back into the body which helps the body to recover quickly.
Higher doses of chemotherapy drugs are more effective in treating tumors but they cause severe damage to bone marrow cells which results in shortage of blood cells which might cause a threat to life.
Possible side effects of high dose chemotherapy
Possible side effects of SCT are generally divided into early and long-term effects.
Short-term, early side effects of high dose chemotherapy are:
- Low blood cell counts (this may be accompanied with fatigue and an increased risk of infection and bleeding)
- Mouth sores
- Diarrhea
- Hair loss
- An increased risk of serious infection
- Nausea and vomiting
- Loss of appetite
- Low red blood cell and platelet counts
Long-term and late side effects of high dose chemotherapy are:
- Damage to the lungs
- Problems with the thyroid or other endocrine glands
- Problems with fertility
- Damage to bones or problems with bone growth (osteoporosis)
- Development of another cancers (such as leukemia) years later
Treatment of Retinoblastoma based on spread of the disease
Treatment options for retinoblastoma depend on the following factors:
- Presence of retinoblastoma in one or both eyes: If retinoblastoma occurs in one or both eyes, the treatment depends on whether the vision can be saved or not.
Many children suffering with retinoblastoma in both eyes are treated with chemotherapy to shrink the tumors, known as chemoreduction, followed by some form of local treatment (possibly radiation therapy).
Surgery (removal of the eye) is recommended for advanced tumors i.e. for those tumors that cannot benefit from chemoreduction and local treatments.
Hereditary form of retinoblastoma increases the probability of development of the disease in the other eye as well.
- The size and location of the tumor in the eyes: If the size of the tumor is too large, it can be reduced with chemotherapy and removed with surgery.
If the size is extremely large and the tumor is located deeply, then the only way to prevent the cancer from spreading is enucleation or surgery.
- The chance for preserving vision in the eyes: For minor retinoblastoma, local treatments such as laser therapy (photocoagulation) or cryotherapy are the only treatments that can save the vision.
If the chances to save the vision are poor and the tumor grows constantly, surgery becomes the only option of treatment.
For tumors which are large or hard-to-treat, a combination of chemotherapy and local treatment is also used.
- The tumor is still confined within the eyes: If retinoblastoma has spread beyond the eye, then strong radiations and chemo-drugs are given to the child to destroy the tumors and sometimes, even the eye is also removed.
If retinoblastoma extends only to the orbit of the eye, a rarely combined treatment of chemotherapy, enucleation , and radiation therapy is often successful.
If retinoblastoma extends from the orbit to distant parts of the body such as the liver or the bones and bone marrow, a stem cell transplant is often a suggested method.
After the treatment of Retinoblastoma
What are the after-effects of treatment for retinoblastoma?
The after-effects are the main concerns for families during the treatment of retinoblastoma. The reoccurrence of retinoblastoma and its metastasis becomes a major point of concern for the patient.
Coping with the treatment is another long-term effect of the treatment of retinoblastoma. The patient should consider the following important points after the treatment of retinoblastoma.
Follow-up care after the treatment
Regular checkups after the treatment of retinoblastoma are very important. It improves the health condition and helps in early detection of a second cancer (if it occurs).
If retinoblastoma affected only one eye and was treated through enucleation, routine eye-exams are conducted for the reoccurrence or metastasis of tumor. The remaining eye is also examined regularly due to an increased risk of occurrence of second retinoblastoma. If the second cancer occurs in eye or any other part of the body, it can be treated early.
Keeping well-maintained medical records after the treatment of retinoblastoma
The medical records of a child or the patient are important because they provide details for the type of treatment which is given to the child (according to the intensity of the disease). If retinoblastoma is inherited then parent’s medical records are also considered.
Genetic counseling and testing for children suffering with retinoblastoma is also an important point for medical studies during and after the treatment.
Medical records also help in defining the extent of the cancer spread and treatment methodology if there is any reoccurrence of the cancer.
Late and Long-Term Effects of Treatment for Retinoblastoma
Most of the children (about 90%) recover from retinoblastoma but managing with it is different for every individual (depending upon the type of cancer and the dose of treatment). The age of a child is also an important factor during and after treatment of retinoblastoma.
The follow-up care and the follow-up treatment both are necessary for retinoblastoma patients. The earlier the problem is diagnosed, the more effective the treatment becomes.
The risk of late and long-term effects depends on certain specific factors such as the age of a child, the dose of treatment, etc. Some after-effects of the treatment of retinoblastoma are stated below:
Eye problems
- Reduced vision in the affected eyes or minor vision changes may cause difficulty in reading, writing and in mobility at the initial stage of tumor development.
- Retinoblastoma may result in loss of sight in one or both eyes.
- Other eye problems are: a child faces some difficulty in judging the distance between objects and difficulty seeing to the side (peripheral vision).
- Eye problems can also lead to changes in balanced vision.
Bone and muscle problems
- After surgery and radiation therapy, the bones around the eye can be deformed.
- The brain becomes deficit of growth hormone due to radiation therapy which can lead to musculoskeleton problems. These problems indicate that the bones and muscles don’t grow as they should, which can lead to underdeveloped muscles.
- If the muscles and bones remain under-developed (such as curvature of the spine, shorter limbs and shorter height), the affected children may require artificial (synthetic) growth hormone for development.
- Children treated with radiation therapy are also at risk for osteoporosis due to low levels of sex and growth hormones.
Kidney problems and heart problems
- The mechanism of kidney can be hindered or kidney cannot function properly due to high dosage of drugs and radiations.
- Kidney problems arise in children after cisplatin is used as a part of chemotherapy and sometimes in stem cell transplant.
- Chemotherapy drugs have greater side effects on the heart. For example, doxorubicin (Adriamycin) weakens heart muscles.
Other body disorders which are the late effects of the treatment of retinoblastoma
- Due to retinoblastoma and its treatments, the growth and development rate lowers down (with a decrease in growth hormone production)
- The sexual ability reduces with time especially due to radiation treatments. As the follicle stimulation hormone and lutineizing hormone level falls, it results in the changes in levels of testosterone in men and estrogen in women
- The reduced levels of thyroid-stimulating hormone TSH (hypothyroidism) leads to thinning of hair, dry skin, constipation, tiredness, weight gain, slower growth of bones, etc.
- Chemotherapy affects the ovaries or testicles in children as they get older and cause problems that lead to puberty, either starting early stage than normal or later than average and also cause infertility.
- There is an increased risk of other cancers (especially in children with hereditary retinoblastoma) such as brain tumors, osteosarcoma ( bone cancer), melanoma, lymphoma, soft tissue sarcomas, etc
- Cisplatin and carboplatin (Paraplatin, Paraplatin AQ), common drugs used in radiotherapy and chemotherapy, can cause loss in hearing ability in young children. This often comes with other problems such as language issues and social development.
Recurrent retinoblastomas
Recurrent cancer simply means that the cancer has come back again after the first treatment. If the cancer returns, there will be another round of tests which are done on a routine basis to learn about the spread of the recurrence of cancer.
There are chances of retinoblastoma to reoccur after the first treatment (especially in case of hereditary retinoblastoma). The treatment methodology depends on the area where the cancer has recurred and the size (or intensity) of the tumor. Possible recommendations can be surgery, radiation therapy, chemotherapy, and/or focal measures, such as photocoagulation, thermotherapy, or cryotherapy after the reoccurrence of retionoblastoma.
Clinical trials are done for recurrent cancer (due to retinoblastoma) and palliative care is provided to the children. Support of family and friends is very important as it motivates the person to live a long healthy happy life.
Related Reads



Latest Posts





ADVERTISEMENT